|
BEPPE Extended explanations |
|
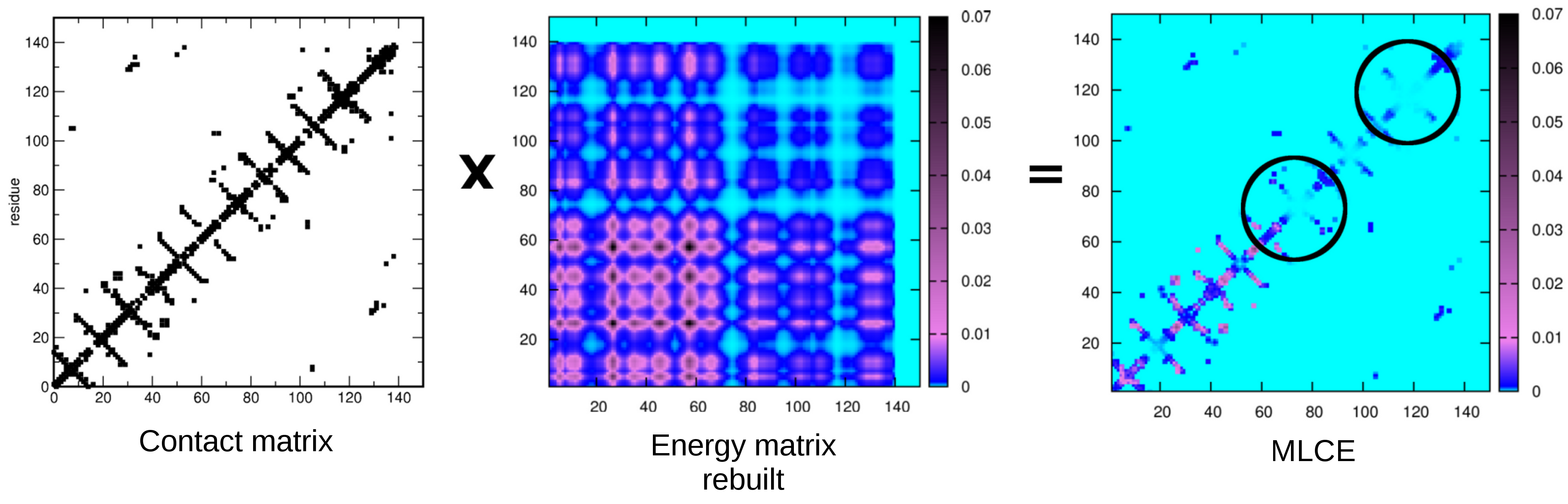
The antibody-binding properties of an antigen depend on its structure and related dynamics. Aiming to predict structural antibody-binding regions of a protein, we investigate an approach based on the integrated analysis of the dynamical and energetic properties of antigens, to identify nonoptimized, low-intensity energetic interaction networks in the protein structure isolated in solution. The method is based on the idea that recognition sites may correspond to localized regions with low-intensity energetic couplings with the rest of the protein, which allows them to undergo conformational changes and to be recognized by a binding partner. Upon analyzing the results on isolated proteins and benchmarking against antibody complexes, it is found that the method successfully identifies binding sites located on the protein surface that are accessible to putative binding partners. The combination of dynamics and energetics can thus discriminate between epitopes and other substructures based only on physical properties. Precisely BEPPE" reads as input a pdb file containing the structure of the protein selected and optimizes it through molecular mechanics steps. On the optimized structure non-bonded energy contributions are calculated for each couple of residues, and a square matrix NxN (where N is the number of amino acids) is built with the energetic values. This matrix is then diagonalized and decomposed in eigenvalues and eigenvectors. The first eigenvector is subsequently used to rebuild an energy matrix multiplying it by its transpose. Afterwardsa contact matrix (whose dimensions are NxN) is build from the protein structure, this matrix contains information on the spacial distance between each couple of residues. The energy matrix rebuilt and the contact matrix are then multiplied through the Hadamard product, the resulting matrix (called Matrix of Local Coupling Energies, MLCE, figure 1) lets to focus on residues forming a patch and showing low couple energetic values, which correspond to sites prone to be subjected to conformational variations upon the binding of a partner like an antibody. For further explanation see material and methods on "Scarabelli G, Morra G, Colombo G. Predicting interaction sites from the energetics of isolated proteins: a new approach to epitope mapping. Biophys J. 2010 May 19;98(9):1966-75." The predicted patches are then aligned through BLAST against the entire human protein sequences available in order to identify possible mimotopes. The segments used for the alignment are taken from the patches and: contains also residues not predicted if they are in the middle of two regions predicted but also not distant in the sequence more than 4 residues from a predicted one Each segment must be at least 5 residues long, otherwise it is not used for the alignment For further explanation on the alignment part see "Amela I, Cedano J, Querol E (2007) Pathogen Proteins Eliciting Antibodies Do Not Share Epitopes with Host Proteins: A Bioinformatics Approach. PLoS ONE 2(6): e512." |